

【储存条件】 室温储存12个月。
【产品简介】
本试剂盒适合于从新鲜或冷冻的动物组织(比如鼠尾、肝脏等),细胞,血液,细菌等多种样品中提取高纯度总DNA。本品可纯化获得分子量最大为50kb的DNA片段,纯化过程不需要使用苯酚或氯仿等有毒溶剂,无需乙醇沉淀。本试剂盒采用优化的缓冲体系使裂解液中的DNA高效特异的结合到硅基质离心吸附柱上,PCR和其他酶促反应的抑制剂可通过两步洗涤步骤被有效去除,最后使用低盐缓冲液或水洗脱,即可得到高纯度DNA。纯化得到的DNA可以直接用于酶切,PCR,Real-Time PCR,文库构建,Southern Blot,分子标记等下游实验。
【自备试剂】 无水乙醇
Enzymatic Lysis Buffer(提取革兰氏阳性菌基因组DNA时须准备)。Enzymatic Lysis Buffer 配方:20 mM Tris,pH 8.0;2 mM Na2-EDTA;1.2%Triton X-100;终浓度为20mg/ml的Lysozyme(溶菌酶)。
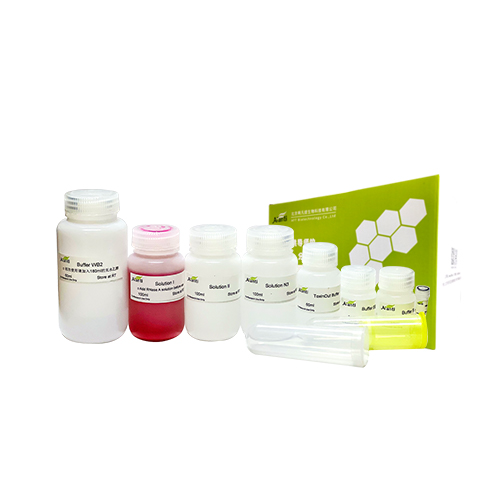
【产品组份】
![]()
![]() 50T
50T
Buffer GTL 15 ml
Buffer GL 15 ml
Buffer GW1(concentrate) 13 ml
Buffer GW2(concentrate) 15 ml
Buffer GE 15 ml
Proteinase K** 1 ml
Spin Columns DM
with Collection Tubes 50个
![]()
** Proteinase K(蛋白酶K):可常温保存,开盖后防止空气及枪头污染,放入4度可以保存更长时间。如果出现絮状物,是因为溶解缓冲液中的钙离子在磷酸盐中可能会产生不溶解性沉淀,离心取上清,不影响使用效果。
【实验前准备及重要注意事项】
1. 样品应避免反复冻融,否则会导致提取DNA片段较小且提取量下降。
2. 如果提取次生代谢产物大量积累或细胞壁厚的细菌培养物的基因组,建议在对数生长期早期收集样品。
3. 第一次使用前应按试剂瓶标签的说明在Buffer GW1和Buffer GW2中加入无水乙醇。
4. 使用前请检查Buffer GTL和Buffer GL是否出现结晶或沉淀,如有结晶或沉淀,请将Buffer GL和Buffer GTL于56˚C水浴重新溶解。
5. 如果下游实验对RNA污染比较敏感,可以在加入Buffer GL前加入4μl DNase-Free的RNase A(100mg/ml),RNase A 本试剂盒并未提供,可单独订购。
【操作步骤】
血液及细胞样本基因组提取
1. 材料处理
a) 如果提取材料为哺乳动物抗凝血液(无核红细胞),可直接向50-200μl新鲜或冷冻的抗凝血液样品中加入Buffer GTL补足至200μl。
b) 如果提取材料为禽类,鸟类,两栖类或更低级生物的抗凝血液,其红细胞为有核细胞,取5-10μl新鲜或冷冻的抗凝血液样品,加入Buffer GTL补足至200μl。
c) 贴壁培养的细胞应先处理为细胞悬液(最大提取量为5x106个细胞),2000 rpm(400xg)离心5分钟,弃尽上清,加200μl GTL,振荡至样品彻底悬浮。
注意:如需去除RNA,可在上述步骤完成后,加入4μl浓度为100mg/ml的RNase A 溶液,涡旋15秒,室温放置2分钟。
2. 加入20μl Proteinase K 溶液,混匀。
3. 加入200μl Buffer GL,涡旋振荡充分混匀,56˚C水浴10分钟。
4. 短暂离心以去除管盖内壁的水珠。加入200μl无水乙醇,涡旋振荡充分混匀,短暂离心。
注意:1)加入Buffer GL和无水乙醇后要立即涡旋震荡混匀。
2)加入Buffer GL和无水乙醇后可能会产生白色沉淀,不会影响后续实验。一些组织在加入BufferGL和无水乙醇后可能形成溶胶状产物,此时推荐进行剧烈震荡或涡旋处理。
5. 上一个步骤中所得溶液全部加入到已装入收集管的吸附柱(Spin Columns DM)中,若一次不能加完溶液,可分多次转入。12,000 rpm(约13,400 xg) 离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
6. 向吸附柱中加入500 μl Buffer GW1(使用前检查是否已加入无水乙醇),12,000 rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
7. 向吸附柱中加入500 μl Buffer GW2(使用前检查是否已加入无水乙醇),12,000 rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
注意:如需进一步提高DNA纯度,可重复步骤7。
8. 12,000 rpm离心2分钟,倒掉收集管中废液。将吸附柱置于室温数分钟,以彻底晾干。
注意:这一步的目的是将吸附柱中残余的乙醇去除,乙醇的残留会影响后续的酶促反应(酶切、PCR等)。
9. 将吸附柱置于一个新的离心管(自备)中,向吸附柱的中间部位悬空加入50-200μl Buffer GE或灭菌水,室温放置2-5分钟,12000rpm离心1分钟,收集DNA溶液,-20˚C保存DNA。
注意:1)如果下游实验对pH值或EDTA敏感,可以用灭菌水洗脱。洗脱液的pH值对洗脱效率有很大影响,若用水做洗脱液应保证其pH值在7.0-8.5(可以用NaOH将水的pH值调到此范围),pH值低于7.0时洗脱效率不高。
2)Buffer GE在65-70˚C水浴预热,离心之前室温孵育5分钟可以增加产量;用另外的50-200 μl BufferGE或灭菌水再次洗脱可以增加产量。
3)如果要提高DNA的终浓度,可将得到的溶液重新加入到吸附柱中,室温放置2-5分钟,12,000 rpm离心1分钟;若洗脱体积小于200 μl,可以增加DNA的终浓度,但可能会减少总产量。如果DNA的量小于1 μg,推荐用50 μl Buffer GE或灭菌水洗脱。
4)因为保存在水中的DNA会受到酸性水解作用的影响,如需长期保存,推荐用Buffer GE洗脱并于-20˚C保存。
动物组织基因组提取
1. 材料处理
如果提取材料为动物组织,取25 mg(脾组织用量应少于10 mg);如果材料为鼠尾,取一段长度为0.4-0.6 cm的大鼠鼠尾或两段长度为0.4-0.6 cm的小鼠鼠尾。
a. 样本进行液氮研磨或切成小块后置于1.5 ml离心管中,加入180 μl Buffer GTL,将不同样品做好标记。
b. 若使用匀浆器处理样本,匀浆前向样本中加入不超过80 μl Buffer GTL,匀浆后加入100 μl Buffer GTL。
注意:1)确保各组织的量不要超出推荐范围。
2)组织样本在加入Buffer GTL之前用液氮研磨或加入Buffer GTL用匀浆器匀浆处理,可以增加裂解效率。
2. 加入20 μl Proteinase K,涡旋震荡使样品彻底混匀。56˚C水浴,直至组织完全裂解,孵育过程中可每隔一段时间颠倒或震荡离心管使样品分散。
注意:1)不同组织消化时间不同,通常1-3小时即可完成,鼠尾需要消化6-8小时,必要时过夜消化,不会影响后续操作。
2)如果孵育和涡旋震荡后仍然有胶状物质,延长56˚C孵育时间或再加入20 μl Proteinase K消化。
3)如需去除RNA,可在上述步骤完成后,加入4μl的浓度为100mg/ml的RNase A溶液,涡旋15秒,室温放置5-10分钟。
3. 加入200 μl Buffer GL,涡旋震荡充分混匀,70˚C水浴10分钟。短暂离心后加入200 μl无水乙醇,涡旋震荡充分混匀。
注意:1)加入Buffer GL和无水乙醇后要立即涡旋震荡混匀。
2)加入Buffer GL和无水乙醇后可能会产生白色沉淀,不会影响后续实验。一些组织(如脾,肺)在加入Buffer GL和无水乙醇后可能形成溶胶状产物,此时推荐进行剧烈震荡或涡旋处理。
4. 短暂离心,将步骤3所得溶液全部加入到已装入收集管的吸附柱(Spin Columns DM)中,若一次不能加完溶液,可分多次转入。12,000 rpm(约13,400xg )离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
5. 向吸附柱中加入500 μl Buffer GW1(使用前检查是否已加入无水乙醇),12,000 rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
6. 向吸附柱中加入500 μl Buffer GW2(使用前检查是否已加入无水乙醇),12,000 rpm。离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
注意:如需进一步提高DNA纯度,可重复步骤6。
7. 12,000 rpm离心2分钟,倒掉收集管中废液。将吸附柱置于室温数分钟,以彻底晾干。
注意:这一步的目的是将吸附柱中残余的乙醇去除,乙醇的残留会影响后续的酶促反应(酶切,PCR等)。
8. 将吸附柱置于一个新的离心管(自备)中,向吸附柱的中间部位悬空加入50-200 μl Buffer GE或灭菌水,室温放置2-5分钟,12,000 rpm离心1分钟,收集DNA溶液,-20℃保存DNA。
注意: 1)如果下游实验对pH值或EDTA敏感,可以用灭菌水洗脱。洗脱液的pH值对洗脱效率有很大影响,若用水做洗脱液应保证其pH值在7.0-8.5(可以用NaOH将水的pH值调到此范围),pH值低于7.0时洗脱效率不高。
2)Buffer GE在65-70°C水浴预热,离心之前室温孵育5分钟可以增加产量;用另外的50-200 μl BufferGE或灭菌水再次洗脱可以增加产量。
3)如果要提高DNA的终浓度,可将得到的溶液重新加入到吸附柱中,室温放置2-5分钟,12,000 rpm离心1分钟;若洗脱体积小于200 μl,可以增加DNA的终浓度,但可能会减少总产量。如果DNA的量小于1 μg,推荐用50 μl Buffer GE或灭菌水洗脱。
4)因为保存在水中的DNA会受到酸性水解作用的影响,如需长期保存,推荐用Buffer GE洗脱并于-20°C保存。
细菌基因组提取
1. 细菌样本预处理
1a. 革兰氏阴性菌
(1) 取细菌培养物1-5 ml(106-108个细胞,最多不超过2×109个细胞)置于离心管(自备)中,12,000 rpm(~13,400xg)离心1分钟,尽量吸净上清。
(2) 向沉淀中加入180 μl Buffer GTL,振荡使菌体重悬。
(3) 加入20 μl Proteinase K,涡旋混匀,56°C孵育,直至菌体完全裂解,孵育过程中每隔一段时间颠倒或震荡离心管使样本分散。
注意:如需去除RNA,可在上述步骤完成后,加入4 μl浓度为100 mg/ml 的RNase A溶液,震荡混匀,室温放置5-10分钟。
(4) 加入200 μl Buffer GL,涡旋震荡混匀。
1b. 革兰氏阳性菌
(1) 取细菌培养物1-5 ml(106-108个细胞,最多不超过2×109个细胞)置于离心管(自备)中,12,000 rpm离心1分钟,尽量吸净上清。
(2) 加入180 μl Enzymatic Lysis Buffer(自备)使菌体重悬。
(3) 37°C孵育30分钟。
(4) 加入20 μl Proteinase K涡旋震荡,充分混匀。加入200 μl Buffer GL,涡旋震荡混匀。56℃孵育30分钟。
注意: 1)如果需要,95°C孵育15分钟可以使病原体失活,但是95°C孵育会造成一些DNA的降解。
2)如需去除RNA,可在上述步骤完成后,加入4 μl浓度为100 mg/ml 的RNase A溶液,震荡混匀,室温放置5-10分钟。
2. 加入200 μl无水乙醇,涡旋震荡充分混匀。
注意:加入无水乙醇后可能会产生白色沉淀,不会影响后续实验。
3. 将步骤2所得溶液(包括形成的沉淀)全部加入到已装入收集管的吸附柱(SpinColumns DM)中,若一次不能加完溶液,可分多次转入。12,000 rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
4. 向吸附柱中加入500 μl Buffer GW1(使用前检查是否已加入无水乙醇),12,000 rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
5. 向吸附柱中加入500 μl Buffer GW2(使用前检查是否已加入无水乙醇),12,000 rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
注意:如需进一步提高DNA纯度,可重复步骤5。
6. 12,000 rpm离心2分钟,倒掉收集管中废液。将吸附柱置于室温数分钟,以彻底晾干。
注意:这一步的目的是将吸附柱中残余的乙醇去除,乙醇的残留会影响后续的酶促反应(酶切、PCR等)。
7. 将吸附柱置于一个新的离心管(自备)中,向吸附柱的中间部位悬空加入50-200 μl Buffer GE或灭菌水,室温放置2-5分钟,12,000 rpm离心1分钟,收集DNA溶液,-20°C保存DNA。
注意: 1)如果下游实验对pH值或EDTA敏感,可以用灭菌水洗脱。洗脱液的pH值对洗脱效率有很大影响,若用水做洗脱液应保证其pH值在7.0-8.5(可以用NaOH将水的pH值调到此范围),pH值低于7.0时洗脱效率不高。
2)Buffer GE在65-70°C水浴预热,离心之前室温孵育5分钟可以增加产量;用另外的50-200 μl Buffer GE或灭菌水再次洗脱可以增加产量。
3)如果要提高DNA的终浓度,可将得到的溶液重新加入到吸附柱中,室温放置2-5分钟,12,000 rpm离心1分钟;若洗脱体积小于200 μl,可以增加DNA的终浓度,但可能会减少总产量。如果DNA的量小于1 μg,推荐用50 μl Buffer GE或灭菌水洗脱。
4)因为保存在水中的DNA会受到酸性水解作用的影响,如需长期保存,推荐用Buffer GE洗脱并于-20℃保存。


